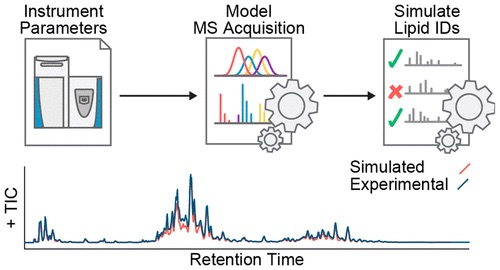
The issue of effectively profiling the diversity and range of biomolecules is an important one to consider in Mass Spectrometry, and relies on well-sought out selection of acquisition parameters. However, acquisition parameters are generally selected in a way that is time-consuming and tends to produce lacking results.
By creating an algorithm which simulates LC-MS/MS lipidomic data acquisition performance in a benchtop quadrupole-Orbitrap Mass Spectrometer system and pairing it with an algorithm that defines constrained parameter optimization, researchers were able to efficiently identify LC-MS/MS method parameter sets for specific sample matrices. Additionally, researchers used a simulation called in silico to demonstrate how developments in mass spectrometer speed and sensitivity will result in even more effective biomolecule identification.